First Edition | 25th May, 2023
The first BEE Meeting was held in Taastrup, Denmark on 25th May, 2023, and served as a pivotal platform for leading experts, clinicians, and patients to gather and share knowledge about Hereditary Hemorrhagic Telangiectasia (HHT).
Read more about this event here.
The key messages from this meeting are summarised below
The clinical diagnostic criteria for HHT were reviewed. Mutation diagnostics are not included in these clinical criteria. In families with a known family mutation, the HHT diagnosis can be confirmed or ruled out for first-degree relatives.
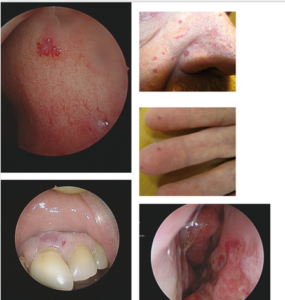
The presence of telangiectatic lesions is part of the Curacao Criteria (2000) (1), though at that time, an exact description of the appearance of the lesions was lacking. Several publications have focused on the distribution and morphology of these lesions (2). There is a general agreement that the number of lesions increases with age (3). The telangiectatic lesions typically occur in the locations mentioned in the Curacao Clinical diagnostic Criteria of HHT: fingers, nose, oral mucosa, skin of the face, and lips.
Telangiectatic lesions can exhibit various appearances. The classic well-known lesions are round, elevated, or flat and can become tortuous or confluent. The color may vary, appearingpinkish or blue. Their morphology and number may differ between HHT subtypes, but not in away that allows the clinician to differentiate between the different HHT subtypes (4).
HHT patients also have elongated linear lesions. Although the linear lesions were discussed as potentially characteristic of HHT, they cannot be considered part of the diagnostic criteria at this point.
Examples of telangiectatic lesions are shown in the figures below.
Bibliography
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ,Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia(Rendu-Osler-Weber syndrome). Am J Med Genet. 2000 Mar 6;91(1):66-7.
- Folz BJ, Lippert BM, Wollstein AC, Tennie J, Happle R, Werner JA. Mucocutaneoustelangiectases of the head and neck in individuals with hereditary hemorrhagictelangiectasia ‐‐ analysis of distribution and symptoms. Eur J Dermatol. 2004;14:407–11.
- Letteboer TG, Mager HJ, Snijder RJ, Lindhout D, Ploos van Amstel H, Zanen P, et al.Genotype‐phenotype relationship for localization and age distribution of telangiectases inhereditary hemorrhagic telangiectasia. Am J Med Genet A. 2008;146a: 2733–9.
- Hyldahl SJ, El-Jaji MQ, Schuster A, Kjeldsen AD. Skin and mucosal telangiectatic lesions inhereditary hemorrhagic telangiectasia patients. Int J Dermatol. 2022 Dec;61(12):1497-1505.doi: 10.1111/ijd.16320. Epub 2022 Jul 6. PMID: 35792874; PMCID: PMC9796122.
HHT follows an autosomal dominant inheritance pattern with significant intra-familial variability and age-dependent penetrance. This genetic disorder is due to pathogenic variants primarily in ENG or ACVRL1, while SMAD4 variants are rare (occurring in around 3% of cases)and lead to HHT combined with juvenile polyposis. In a smaller fraction of HHT families, the pathogenic gene variant remains unidentified. Genetic testing for HHT is recommended to encompass relevant differential diagnoses (RASA1, EPHB4, GDF2/BMP9) in addition to ENG,ACVRL1, and SMAD4.
Prenatal and preimplantation genetic testing are possible if the pathogenic variant is known inthe family, although it is seldom requested by HHT patients (especially in ENG and ACVRL1families). Decisions regarding prenatal genetic testing rest with the parents, but thoroughdiscussion of all related issues is advisable.
The two-hit hypothesis has been widely accepted for many years as a plausible explanation for the observed phenotypic variability in HHT. The germline heterozygous pathogenic variant(first hit) result in monoallelic protein loss in endothelial cells. The second hit is thought to be either an environmental stimulus (such as inflammation, hypoxia, vascular injury, or trauma) or a local genetic variant on the normal HHT allele (of the same gene), or possibly a combination of both. In all cases, the consequence is impaired endothelial cell function, leading to the development of telangiectases and AVMs. Current clinical data, as well as in vivo and in vitro experimental results, support the second-hit hypothesis as an explanation for the incomplete penetrance and significant clinical variation observed in HHT. Nevertheless, substantial gaps in our knowledge persist.